









Spis treści
Choroba Hirschsprunga
Co to jest ?
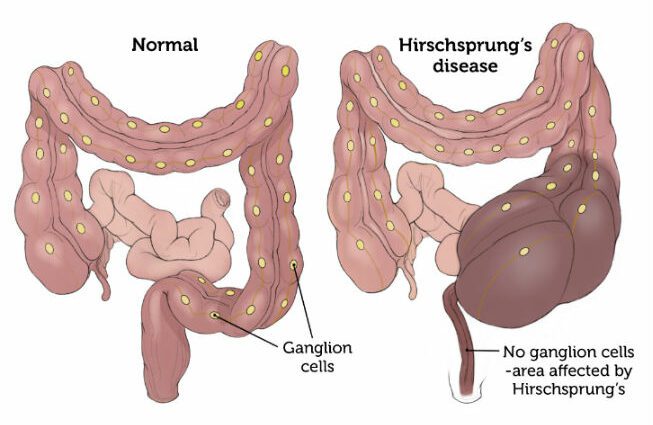
Choroba Hirschsprunga (HSCR) charakteryzuje się porażeniem końcowej części jelita grubego.
Ta patologia pojawia się od urodzenia i jest konsekwencją braku zwojów nerwowych (komórek tworzących wybrzuszenie na ścieżce nerwu) w ścianie jelita.
Połykanie pokarmu przez przewód pokarmowy aż do jego wydalenia jest w dużej mierze możliwe dzięki perystaltyce jelit. Ta perystaltyka to zespół skurczów mięśni jelit, który umożliwia posuwanie się bolusu pokarmowego wzdłuż przewodu pokarmowego.
W tej sytuacji, gdy nie ma zwojów nerwowych w jelicie grubym, organizm nie zapewnia perystaltyki. W tym sensie powstaje rozszerzenie jelita i zwiększenie jego objętości.
Związane z tym objawy są tym ważniejsze, jeśli obszar zwojów nerwowych jest duży. (1)
Choroba ta jest zatem definiowana przez nietypowe objawy jelitowe: niedrożność jelit. Jest to blokada przepływu i gazu prowadząca do bólu brzucha, kolki (skurcze jelit), nudności, wzdęć itp.
HSCR dotyka około 1 na 5 urodzeń rocznie. Postać dotykająca końcową część okrężnicy (jelito grube) dotyka głównie chłopców. (000) Dziewczęta są bardziej narażone na rozwój tej choroby w bardziej rozpowszechnionej formie. (2)
Ta patologia dotyczy głównie niemowląt i małych dzieci. (3)
Wykazano kilka postaci choroby (2):
– kształt „klasyczny” lub zwany też „krótkosegmentowym”. Ta forma występuje najczęściej u pacjentów z tą patologią, do 80%. Ta postać choroby dotyczy końcowej części okrężnicy do odcinka odbytniczego;
– forma „długoodcinkowa”, która rozciąga się na esicy, dotyczy prawie 15% pacjentów;
– forma „kolki całkowitej”, obejmująca okrężnicę jako całość, dotyczy 5% pacjentów.
objawy
Pasaż jelitowy jest kontrolowany przez układ nerwowy. Zwoje nerwowe znajdują się zatem w jelicie, umożliwiając przekazywanie informacji z mózgu w celu kontrolowania perystaltyki jelit, a tym samym postępu pokarmu w przewodzie pokarmowym.
Brak tych węzłów, w przypadku choroby Hirschsprunga, uniemożliwia przekazywanie informacji, a tym samym blokuje perystaltykę jelit. Pokarm nie może już przejść przez jelita i zostaje zablokowany w przewodzie pokarmowym.
Objawy tej choroby są zwykle zauważalne bardzo wcześnie po urodzeniu. Jednak w niektórych przypadkach mogą pojawić się po roku lub dwóch latach. (3)
Objawy występujące u noworodków i dzieci to głównie:
– trudności tranzytowe;
– niemożność wydalenia smółki (pierwszych odchodów noworodka) w ciągu pierwszych 48 godzin;
- zaparcie;
– żółtaczka;
– wymioty ;
- biegunka;
- ból brzucha;
– niedożywienie.
Objawy dotykające starsze dzieci to:
– ciężkie zaparcia z powikłaniami (brak wzrostu i masy ciała);
– złe odżywianie;
– wzdęcie brzucha;
- gorączka.
Dziecko może również rozwinąć infekcje jelitowe, takie jak zapalenie jelit.
Mogą być również widoczne dodatkowe nieprawidłowości: niedosłuch czuciowo-nerwowy (zespół Waardenburga-Shah), niepełnosprawność intelektualna (zespół Mowata-Wilsona), centralna hipowentylacja pęcherzykowa (zespół Haddada), nieprawidłowości kończyn (zespół Bardeta) Biedla), rak rdzeniasty tarczycy (zespół wielowydzielniczy). neoplazja typu 2B) lub nieprawidłowości chromosomalne (zespół Downa). (2)
Początki choroby
Choroba Hirschsprunga jest spowodowana nieprawidłowościami w rozwoju jelitowego układu nerwowego. Jest to aganglionoza, czyli brak zwojów nerwowych (zwanych również „komórkami Cajala”) w jelitach. Ten deficyt węzłów chłonnych jest szczególnie zlokalizowany w końcowej części jelita grubego (okrężnicy).
U podmiotu dotkniętego tą patologią ta część jelita pozostaje zatem w stanie tonicznego i trwałego skurczu. Ta sytuacja prowadzi do niedrożności jelit. (2)
W rozwój choroby Hirschsprunga zaangażowane są zarówno czynniki genetyczne, jak i środowiskowe. (2)
Rzeczywiście, pewne geny zostały wykazane w rozwoju tej patogenezy. Jest to choroba poligenetyczna, która dotyczy w szczególności genów:
- Prokoonkogen ret (RET);
– gen czynnika neutrotroficznego pochodzenia glejowego (GDNF);
– gen receptora endoteliny typu B (EDNRB);
– gen endoteliny 3 (EDN3);
– gen enzymu konwertującego endotelinę 1 (ECE1);
– gen cząsteczki adhezyjnej komórki L1 (L1CAM).
Czynniki ryzyka
Jak wspomniano wcześniej, choroba Hirschsprunga jest konsekwencją braku zwojów nerwowych w jelicie grubym aż do odbytu, co zapobiega perystaltyce jelit, a tym samym wznoszenia się pokarmu do tego poziomu.
Ten deficyt komórek Cajala (zwojów nerwowych) jest konsekwencją deficytu wzrostu tych komórek podczas rozwoju płodowego. Przyczyny tego braku wzrostu komórek przed urodzeniem nie są jeszcze znane. Niemniej jednak wysunięto możliwość związku między ogólnym stanem zdrowia matki w okresie ciąży a brakiem tego typu komórek u płodu.
W rozwoju choroby wykazano wiele genów. Obecność tych genów może być częsta w tej samej rodzinie. Część dziedziczności byłaby wtedy źródłem rozwoju tej choroby.
Ponadto niektóre patologie mogą być dodatkowym czynnikiem ryzyka rozwoju choroby Hirschsprunga. Dzieje się tak szczególnie w przypadku zespołu Downa. (3)
Zapobieganie i leczenie
Diagnozę różnicową przeprowadza się na podstawie charakterystycznych objawów choroby przedstawionych przez badanego: niedrożność jelit, zwężenie odbytu, guzy miednicy itp. (2)
Diagnozę najczęściej związaną z chorobą stawia się na podstawie biopsji odbytnicy. Ta biopsja wykazuje obecność lub brak zwojów nerwowych w jelicie grubym. Ponadto nadekspresja acetylocholiny esterazy (enzymu umożliwiającego hydrolizę acetylocholiny do kwasu octowego i choliny). (2)
W diagnostyce tej patologii można również wykonać lewatywę z baru (badanie rentgenowskie w celu uwidocznienia jelita grubego). Ta metoda umożliwia wizualizację przejściowego obszaru braku komórek nerwowych, wskazującego na rozwój choroby Hischsprunga. Jednak ta technika diagnostyczna nie jest w 100% niezawodna. Rzeczywiście, 10 do 15% przypadków choroby Hirschsprunga nie zostałoby zdiagnozowanych po tej próbie diagnostycznej. (4)
Najważniejszym sposobem leczenia choroby jest operacja. Umożliwia ablację części jelita z niedoborem komórek nerwowych. (4)
W przypadku całkowitego uszkodzenia okrężnicy może być konieczny przeszczep okrężnicy. (2)
Następnie można wykonać stomię (technika chirurgiczna pozwalająca na połączenie dwóch narządów) w celu połączenia operowanej części jelita z odbytem lub z górną częścią jelita. Ta stomia może być stała lub tymczasowa, w zależności od przypadku. (4)
Chirurgia pomaga zmniejszyć objawy związane z chorobą. Rokowanie nie jest jednak pełne i mogą pojawić się powikłania zapalne, które mogą być śmiertelne.