









Spis treści
Jednym z najbardziej charakterystycznych objawów choroby jest patologiczne nagromadzenie miedzi w okolicy różnych narządów, uszkodzenie tkanek, zwłaszcza wątroby, problemy układu nerwowego, zmiany w tęczówce oka.
Co to jest choroba Wilsona-Konowałowa
Termin choroba Wilsona-Konowałowa jest dziedziczną patologią. Występuje, gdy rodzice przekazują dziecku wadliwy gen (ATP7B). Stan ten dotyczy patologii autosomalnych recesywnych, to znaczy występuje, gdy każdy z rodziców nosi w swoich komórkach podobny gen, a dziecko dziedziczy oba geny jednocześnie – od matki i od ojca.
Ten wadliwy gen daje instrukcje dotyczące syntezy białka, które reguluje wymianę i transport miedzi w organizmie. Z powodu swojej wady miedź gromadzi się w wątrobie, koncentruje się w zwojach nerwowych i odkłada się w tęczówce oka. Patologia nie jest powszechna, czasami bardzo trudno ją rozpoznać, zwłaszcza jeśli w rodzinie nie ma takich pacjentów.
Przyczyny choroby Wilsona-Konowałowa u dorosłych
Kluczowym procesem w tej patologii jest dziedziczenie wadliwego genu od rodziców. Znajduje się na 13. chromosomie i reguluje metabolizm miedzi.
Ciało dorosłego człowieka zawiera średnio 50-70 mg miedzi i potrzebuje nie więcej niż 2 mg tego pierwiastka dziennie, który pochodzi z pożywienia.
Ogromna większość mikroelementów (95%) jest przenoszona w ścisłym związku z białkiem osocza, ceruloplazminą. Jest stale tworzona przez wątrobę, a tylko około 5% miedzi jest transportowane wraz z albuminą.
Miedź jest potrzebna do udziału w procesach metabolicznych, w tym oksydacyjnych. Jeśli rozwinie się choroba Wilsona, jej wydalanie zostaje zakłócone, wzrasta stężenie w osoczu, skąd rozprzestrzenia się do tkanek. Główna akumulacja miedzi występuje w mózgu, w okolicy tęczówki, w wątrobie, a także w nerkach. Nadmiar mikroelementu działa toksycznie.
Objawy choroby Wilsona-Konowałowa u dorosłych
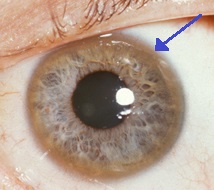
Możliwe przejawy są bardzo zróżnicowane. Najczęściej cierpi wątroba (około 40 – 50% przypadków), w innych przypadkach można zauważyć zmiany neurologiczne i problemy psychiczne. Przy uszkodzeniu układu nerwowego i wzroku pojawia się typowy objaw – manifestacja pierścienia Kaisera-Fleischera (występuje na skutek odkładania się miedzi w tęczówce ze specyficznym brązowym zabarwieniem).
W brzusznej postaci choroby objawy zwykle pojawiają się bliżej wieku 40 lat. Kluczowe cechy to:
- marskość wątroby;
- przewlekłe lub piorunujące (piorunujące) zapalenie wątroby.
W dzieciństwie częściej występuje sztywna arytmohiperkinetyczna odmiana choroby. Rozpoczyna się sztywnością (zagęszczenie, słaba podatność) mięśni, zaburzeniami wyrazu twarzy, zaburzeniami mowy, problemami z wykonywaniem ruchów wymagających małej motoryki i pewnym spadkiem inteligencji. Choroba postępuje stopniowo, z okresami zaostrzeń i remisji.
Wariant drżenia choroby Wilsona zwykle występuje w wieku od 10 do 30 do 35 lat. Mogą wystąpić takie objawy jak drżenie, spowolnienie ruchów, opóźnienie mowy, napady padaczkowe, problemy psychiczne.
Najrzadszą postacią choroby są zaburzenia pozapiramidowo-korowe. Podobnie jest ze wszystkimi postaciami, dodatkowo będą napady konwulsyjne, poważne problemy intelektualne, zaburzenia ruchu.
Leczenie choroby Wilsona-Konowałowa u dorosłych
Wczesna diagnoza jest niezbędna do skutecznego leczenia. Nie zawsze jest to łatwe, szczególnie w sytuacjach, gdy nie ma typowych objawów i zmian tęczówki z pojawieniem się pierścienia. Najczęściej pacjenci przychodzą do neurologa, gastroenterologa lub problem wykrywa okulista.
Diagnostyka
Jeśli mówimy o manifestacji objawów ocznych, lekarz najpierw bada stan oczu za pomocą lampy szczelinowej, aby potwierdzić obecność pierścienia Kaiser-Fleischer.
Pokazano wyznaczenie testów biochemicznych krwi i moczu, które wykażą zwiększoną zawartość miedzi w moczu i zmniejszone stężenie ceruloplazminy w osoczu krwi.
CT lub MRI pokażą procesy zanikowe w mózgu i móżdżku, uszkodzenie jąder podstawnych.
Dodatkowo przeprowadzana jest konsultacja z genetykiem oraz szereg badań genetycznych identyfikujących wadliwe geny.
Nowoczesne zabiegi
Główną metodą leczenia tej choroby jest wyznaczenie leków tiolowych, zwłaszcza unitiolu lub D-penicylamina, cuprenil. Leki są przyjmowane przez długi czas, lekarz dobiera najbardziej optymalną dawkę, która pozwoli uniknąć skutków ubocznych.
Dodatkowo lekarz może stosować leki z grupy neuroleptyków, ze sztywnością mięśni – lewodopę lub karbidopę.
W ciężkich przypadkach wskazane jest przeszczepienie wątroby i leczenie immunosupresyjne. Możliwe jest zastosowanie biohemoperfuzji z izolatem żywych elementów komórkowych śledziony z wątrobą.
Dodatkowo konieczne jest przestrzeganie diety z wyjątkiem pokarmów zawierających duże ilości miedzi.
Zapobieganie chorobie Wilsona-Konowałowa u dorosłych w domu
„W profilaktyce patologii” – mówi. neurolog Valentina Kuzmina, – konieczne jest przestrzeganie diety nr 5, a także ograniczenie spożycia miedzi do 1 g dziennie – wyklucz orzechy, suszone owoce, czekoladę, raki, herbatniki, pełnoziarniste. Zaleca się również przyjmowanie leków z grupy witaminy B6, unitiolu, trientyny.
Popularne pytania i odpowiedzi
Rozmawialiśmy o problemach choroby Wilsona-Konowałowa, jej powikłaniach i możliwości samoleczenia neurolog Valentina Kuzmina.
● uszkodzenie wątroby, zwłaszcza jeśli rozwinie się marskość wątroby;
● choroba psychiczna – znaczne upośledzenie umysłowe, psychoza;
● choroby neurologiczne – zaburzona koordynacja, w której występuje również drżenie kończyn, zaburzenia chodu, zwiększone wydzielanie śliny.