









Spis treści
Progeria lub zespół Hutchinsona-Gilforda
Progeria to rzadka choroba genetyczna charakteryzująca się wczesnym starzeniem się dziecka.
Definicja progerii
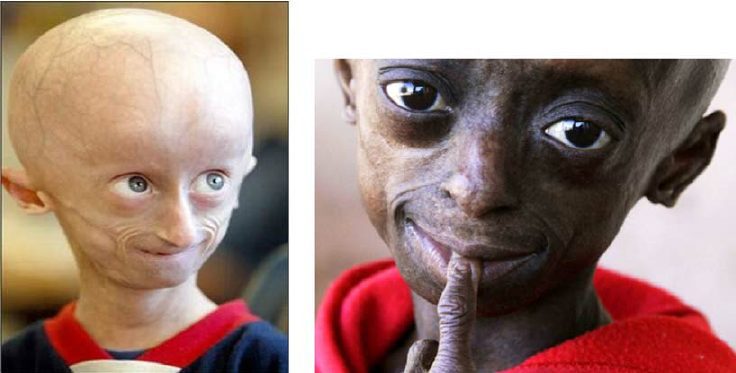
Progeria, znana również jako zespół Hutchinsona-Gilforda, jest rzadkim zaburzeniem genetycznym. Charakteryzuje się zwiększonym starzeniem się organizmu. Dzieci z tą patologią wykazują wczesne oznaki starości.
Po urodzeniu dziecko nie wykazuje żadnych nieprawidłowości. Wydaje się „normalne” aż do niemowlęctwa. Stopniowo jego morfologia i ciało rozwijają się nienormalnie: rośnie wolniej niż normalnie i nie przybiera na wadze dziecka w jego wieku. Na pierwszy rzut oka rozwój jest również opóźniony. Przedstawia wyłupiaste oczy (z reliefem, mocno wysunięte), bardzo cienki i haczykowaty nos, cienkie wargi, mały podbródek i odstające uszy. Progeria jest również przyczyną znacznego wypadania włosów (łysienie), starzenia się skóry, niedoborów stawów, a nawet utraty podskórnej tkanki tłuszczowej (tłuszczu podskórnego).
Na ogół nie ma to wpływu na rozwój poznawczy i intelektualny dziecka. Jest to w istocie niedobór i konsekwencje dla rozwoju motorycznego, powodujące trudności w siedzeniu, staniu, a nawet chodzeniu.
Pacjenci z zespołem Hutchinsona-Gilforda mają również zwężenie niektórych tętnic, co prowadzi do rozwoju miażdżycy (zablokowania tętnic). Jednak miażdżyca jest głównym czynnikiem zwiększającym ryzyko zawału serca, a nawet udaru mózgu.
Częstość występowania tej choroby (liczba dotkniętych nią osób w całej populacji) wynosi 1/4 miliona noworodków na całym świecie. Jest to zatem choroba rzadka.
Ponieważ progeria jest autosomalną dominującą chorobą dziedziczną, osoby najbardziej narażone na rozwój takiej choroby to te, których rodzice również mają progerię. Możliwe jest również ryzyko losowych mutacji genetycznych. Progeria może wtedy dotknąć każdą osobę, nawet jeśli choroba nie występuje w kręgu rodzinnym.
Przyczyny progerii
Progeria jest rzadką i genetyczną chorobą. Przyczyną tej patologii są mutacje w genie LMNA. Gen ten jest odpowiedzialny za tworzenie białka: Lamine A. Ta ostatnia odgrywa ważną rolę w tworzeniu jądra komórkowego. Jest niezbędnym elementem w tworzeniu otoczki jądrowej (błony otaczającej jądro komórek).
Mutacje genetyczne w tym genie prowadzą do nieprawidłowego tworzenia Lamine A. To nieprawidłowo utworzone białko jest przyczyną niestabilności jądra komórkowego, a także wczesnej śmierci komórek organizmu.
Naukowcy zadają sobie obecnie pytanie na ten temat, aby uzyskać więcej szczegółów na temat udziału tego białka w rozwoju organizmu człowieka.
Przenoszenie genetyczne zespołu Hutchinsona-Gilforda następuje poprzez dziedziczenie autosomalne dominujące. Do rozwoju choroby u dziecka wystarczy przekazanie tylko jednej z dwóch kopii określonego genu (od matki lub od ojca).
Co więcej, przypadkowe mutacje (nie wynikające z transmisji genów rodzicielskich) genu LMNA mogą być również przyczyną takiej choroby.
Objawy progerii
Ogólne objawy progerii charakteryzują się:
- wczesne starzenie się organizmu (od dzieciństwa);
- mniejszy przyrost masy ciała niż normalnie;
- mniejszy rozmiar dziecka;
- nieprawidłowości twarzy: cienki, haczykowaty nos, wyłupiaste oczy, mały podbródek, odstające uszy itp.;
- opóźnienia motoryczne, powodujące trudności w staniu, siedzeniu, a nawet chodzeniu;
- zwężenie tętnic, główny czynnik ryzyka miażdżycy.
Czynniki ryzyka progerii
Ponieważ progeria jest rzadką, genetyczną i dziedziczną chorobą autosomalną dominującą, obecność choroby u jednego z obojga rodziców jest najważniejszym czynnikiem ryzyka.
Jakie leczenie progerii?
Objawy związane z progerią są postępujące i mogą prowadzić do śmierci pacjenta.
Obecnie nie jest dostępne żadne lekarstwo na tę chorobę. Jedynym sposobem leczenia progerii jest postępowanie z objawami.
W centrum uwagi znajdują się zatem badania mające na celu wynalezienie metody leczenia takiej choroby.