









Choroba Wilsona
Co to jest ?
Choroba Wilsona jest dziedziczną chorobą genetyczną, która uniemożliwia eliminację miedzi z organizmu. Nagromadzenie miedzi w wątrobie i mózgu powoduje problemy wątrobowe lub neurologiczne. Częstość występowania choroby Wilsona jest bardzo niska, około 1 na 30 osób. (000) Istnieje skuteczne leczenie tej choroby, ale jej wczesna diagnoza jest problematyczna, ponieważ przez długi czas pozostaje niema.
objawy
Nagromadzenie miedzi zaczyna się od urodzenia, ale pierwsze objawy choroby Wilsona często pojawiają się dopiero w okresie dojrzewania lub dorosłości. Mogą być bardzo zróżnicowane, ponieważ nagromadzenie miedzi wpływa na kilka narządów: serce, nerki, oczy, krew… Pierwsze oznaki są wątrobowe lub neurologiczne w trzech czwartych przypadków (odpowiednio 40% i 35%), ale mogą być również psychiatrycznym, nerkowym, hematologicznym i endokrynologicznym. Szczególnie dotknięte są wątroba i mózg, ponieważ w naturalny sposób zawierają najwięcej miedzi. (2)
- Zaburzenia wątroby: żółtaczka, marskość wątroby, niewydolność wątroby…
- Zaburzenia neurologiczne: depresja, zaburzenia zachowania, trudności w uczeniu się, trudności w wyrażaniu siebie, drżenie, skurcze i przykurcze (dystonia)…
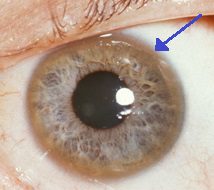
Pierścień Keysera-Fleishera, który otacza tęczówkę, jest charakterystyczny dla gromadzenia się miedzi w oku. Oprócz tych ostrych objawów choroba Wilsona może objawiać się nietypowymi objawami, takimi jak ogólne zmęczenie, ból brzucha, wymioty i utrata masy ciała, anemia i ból stawów.
Początki choroby
Przyczyną choroby Wilsona jest mutacja w genie ATP7B zlokalizowanym na chromosomie 13, który bierze udział w metabolizmie miedzi. Kontroluje produkcję białka ATPazy 2, które odgrywa rolę w transporcie miedzi z wątroby do innych części ciała. Miedź jest niezbędnym budulcem wielu funkcji komórek, ale w nadmiarze miedzi staje się toksyczna i uszkadza tkanki i narządy.
Czynniki ryzyka
Przenoszenie choroby Wilsona jest autosomalne recesywne. Dlatego do rozwoju choroby konieczne jest otrzymanie dwóch kopii zmutowanego genu (od ojca i matki). Oznacza to, że mężczyźni i kobiety są w równym stopniu narażeni, a dwoje rodziców noszących zmutowany gen, ale nie chorzy, ma ryzyko przeniesienia choroby na czterech przy każdym urodzeniu.
Zapobieganie i leczenie
Istnieje skuteczna terapia, która powstrzyma postęp choroby i złagodzi, a nawet zlikwiduje jej objawy. Konieczne jest również jej wczesne rozpoczęcie, ale często mija wiele miesięcy od pojawienia się objawów, aby zdiagnozować tę niemą chorobę, mało znaną i której objawy wskazują na wiele innych stanów (zapalenie wątroby, z powodu którego jest uszkodzenie wątroby i depresja z powodu zaangażowania psychiatrycznego). .
Zabieg „chelatujący” umożliwia przyciągnięcie miedzi i eliminację jej z moczem, ograniczając w ten sposób jej gromadzenie w narządach. Opiera się na D-penicylaminie lub Trientine, lekach przyjmowanych doustnie. Są skuteczne, ale mogą powodować poważne skutki uboczne (uszkodzenie nerek, reakcje alergiczne itp.). Kiedy te efekty uboczne są zbyt istotne, uciekamy się do podawania cynku, który ograniczy wchłanianie miedzi przez jelita.
Przeszczep wątroby może być konieczny, gdy wątroba jest zbyt uszkodzona, co ma miejsce w przypadku 5% osób z zespołem Wilsona (1).
Rodzeństwo osoby dotkniętej chorobą oferuje genetyczne badanie przesiewowe. Daje podstawę do skutecznego leczenia zapobiegawczego w przypadku wykrycia nieprawidłowości genetycznej w genie ATP7B.